Kent Hovind, a former high school science teacher turned creation science evangelist, said that evolution is the "dumbest and most dangerous theory on planet Earth" at a program in the Union on Dec. 6.
Posted on 12/17/2005 3:58:48 AM PST by PatrickHenry
A former high school science teacher turned creation science evangelist told an audience at the University of Wisconsin-Milwaukee last Tuesday that evolution is the “dumbest and most dangerous theory on planet Earth.”
Kent Hovind, founder of Creation Science Evangelism, presented “Creation or Evolution … Which Has More Merit?” to a standing-room only audience in the Union Ballroom on Dec. 6. The event was sponsored by the Apologetics Association, the organization that brought Baptist minister Tim Wilkins to UWM to speak about homosexuality in October.
Members of the Apologetics Association (AA) contacted biology, chemistry and geology professors at UWM and throughout the UW System, inviting them to debate Hovind for an honorarium of $200 to be provided to the individual or group of individuals who agreed.
Before the event began, the “No-Debater List,” which was comprised of slides listing the names of UWM science professors who declined the invitation, was projected behind the stage.
Dustin Wales, AA president, said it was his “biggest disappointment” that no professor agreed to debate Hovind.
“No professor wanted to defend his side,” he said. “I mean, we had seats reserved for their people … ’cause I know one objection could have been ‘Oh, it’s just a bunch of Christians.’ So we had seats reserved for them to bring people to make sure that it’s somewhat more equal, not just all against one. And still nobody would do it.”
Biology professor Andrew Petto said: “It is a pernicious lie that the Apologetics (Association) is spreading that no one responded to the challenge. Many of us (professors) did respond to the challenge; what we responded was, ‘No, thank you.’ ”
Petto, who has attended three of Hovind’s “performances,” said that because Hovind presents “misinterpretations, half truths and outright lies,” professors at UWM decided not to accept his invitation to a debate.
“In a nutshell, debates like this do not settle issues of scientific understanding,” he said. “Hovind and his arguments are not even in the same galaxy as legitimate scientific discourse. This is why the faculty here has universally decided not to engage Hovind. The result would be to give the appearance of a controversy where none exists.”
He added, “The faculty on campus is under no obligation to waste its time supporting Hovind’s little charade.”
Hovind, however, is used to being turned down. Near the end of his speech, he said, “Over 3,000 professors have refused to debate me. Why? Because I’m not afraid of them.”
Hovind began his multimedia presentation by asserting that evolution is the “dumbest and most dangerous” theory used in the scientific community, but that he is not opposed to science.
“Our ministry is not against science, but against using lies to prove things,” he said. He followed this statement by citing biblical references to lies, which were projected onto screens behind him.
Hovind said: “I am not trying to get evolution out of schools or to get creation in. We are trying to get lies out of textbooks.” He added that if removing “lies” from textbooks leaves no evidence for evolutionists’ theory, then they should “get a new theory.”
He cited numerous state statutes that require that textbooks be accurate and up-to-date, but said these laws are clearly not enforced because the textbooks are filled with lies and are being taught to students.
Petto said it is inevitable that textbooks will contain some errors.
“Sometimes, this is an oversight. Sometimes it is the result of the editorial and revision process. Sometimes it is the result of trying to portray a rich and complex idea in a very few words,” he said.
The first “lie” Hovind presented concerned the formation of the Grand Canyon. He said that two people can look at the canyon. The person who believes in evolution would say, “Wow, look what the Colorado River did for millions and millions of years.” The “Bible-believing Christian” would say, “Wow, look what the flood did in about 30 minutes.”
To elaborate, Hovind discussed the geologic column — the chronologic arrangement of rock from oldest to youngest in which boundaries between different eras are marked by a change in the fossil record. He explained that it does not take millions of years to form layers of sedimentary rock.
“You can get a jar of mud out of your yard, put some water in it, shake it up, set it down, and it will settle out into layers for you,” he said. Hovind used this concept of hydrologic sorting to argue that the biblical flood is what was responsible for the formation of the Grand Canyon’s layers of sedimentary rock.
Hovind also criticized the concept of “micro-evolution,” or evolution on a small, species-level scale. He said that micro-evolution is, in fact, scientific, observable and testable. But, he said, it is also scriptural, as the Bible says, “They bring forth after his kind.”
Therefore, according to the Bible and micro-evolution, dogs produce a variety of dogs and they all have a common ancestor — a dog.
Hovind said, however, Charles Darwin made a “giant leap of faith and logic” from observing micro-evolution into believing in macro-evolution, or evolution above the species level. Hovind said that according to macro-evolution, birds and bananas are related if one goes back far enough in time, and “the ancestor ultimately was a rock.”
He concluded his speech by encouraging students to personally remove the lies from their textbooks and parents to lobby their school board for accurate textbooks.
“Tear that page out of your book,” he said. “Would you leave that in there just to lie to the kids?”
Petto said Hovind believes the information in textbooks to be “lies” because his determination is grounded in faith, not science.
“Make no mistake, this is not a determination made on the scientific evidence, but one in which he has decided on the basis of faith alone that the Bible is correct, and if the Bible is correct, then science must be wrong,” he said.
Petto said Hovind misinterprets scientific information and then argues against his misinterpretation.
“That is, of course, known as the ‘straw man’ argument — great debating strategy, but nothing to do with what scientists actually say or do,” he said. “The bottom line here is that the science is irrelevant to his conclusions.”
Another criticism of Hovind’s presentation is his citation of pre-college textbooks. Following the event, an audience member said, “I don’t think using examples of grade school and high school biology can stand up to evolution.”
Petto called this an “interesting and effective rhetorical strategy” and explained that Hovind is not arguing against science, but the “textbook version” of science.
“The texts are not presenting the research results of the scientific community per se, but digesting and paraphrasing it in a way to make it more effective in learning science,” he said. “So, what (Hovind) is complaining about is not what science says, but what the textbooks say that science says.”
Petto said this abbreviated version of scientific research is due, in part, to the editorial and production processes, which impose specific limits on what is included.
He added that grade school and high school textbooks tend to contain very general information about evolution and pressure from anti-evolutionists has weakened evolutionary discussion in textbooks.
“Lower-level texts … tend to be more general in their discussions of evolution and speak more vaguely of ‘change over time’ and adaptation and so on,” he said. “Due to pressure by anti-evolutionists, textbook publishers tend to shy away from being ‘too evolutionary’ in their texts … The more pressure there is on schools and publishers, the weaker the evolution gets, and the weaker it gets, the more likely that it will not do a good job of representing the current consensus among biologists.”
Hovind has a “standing offer” of $250,000 for “anyone who can give any empirical evidence (scientific proof) for evolution.” According to Hovind’s Web site, the offer “demonstrates that the hypothesis of evolution is nothing more than a religious belief.”
The Web site, www.drdino.com, says, “Persons wishing to collect the $250,000 may submit their evidence in writing or schedule time for a public presentation. A committee of trained scientists will provide peer review of the evidence offered and, to the best of their ability, will be fair and honest in their evaluation and judgment as to the validity of the evidence presented.”
Wales said the AA’s goal in bringing Hovind to UWM was “to crack the issue on campus” and bring attention to the fallibility of evolution.
“The ultimate goal was to say that, ‘Gosh, evolution isn’t as concrete as you say it is, and why do you get to teach everyone this non-concrete thing and then not defend it when someone comes and says your wrong?’ ” he said. “It’s just absurd.”
"My grampa was no monkey."
(I stole this paraphrase from another FReeper)
I will have to read that link about the various Christian religions tomorrow...
But what is interesting on the religious threads on FR, is that you will have one poster who belongs to one religion telling another poster who belongs to another religion tell one another, not only are they wrong about their own interpretations of Biblical passages, they are also not even 'Christians'...imagine that...self professed Christians telling other self professed Christians of another religion that they dont even qualify as being a Christian...its gets to mixed up and circular after a while, that one can hardly tell the players without signs on their backs denoting what religion they approve of and what religion they disapprove of...its a mess...
So, when you read things like that you realize its no wonder Christianity is fragmented...evolution has nothing at all to do with it...its all the various 'Christian religions', making the claim, that they are the one and only road to salvation, that have fragmented Christianity..
Just like God is powerful and loving enough to create homosexuals. Incredible!
The ability of unbelievers to twist God's Word to fit their unbelief is amazing.
One of my favorite examples involved the threads that utterly savaged anyone who said anything less than 100% complimentary about the film, "The Passion of the Christ". Check out this post of mine on one of those threads, excerpting a couple of dozen disgusting attacks on an author who had merely pointed out that Mel Gibson had missed a chance to reassure his critics during an interview, and had instead said some things which were likely to keep the controversy alive.
The author was immediately denounced as "an angry socialist as full of the aggressive zeal possessed by any loyal Trotskyite", who was "beneath contempt", who "probably stood up for Clinton", who "believes other people's suffering has less significance", a "rancid dingbat" and "lying leftist", etc. etc. A quick Google of her other essays, however, revealed positions which would not be out of place in a Mark Steyn or Ann Coulter column... (See link for details).
It was bizarre to see how many people jumped to vicious conclusions about the author based on, well, nothing at all.
Where did you go to school and what did you study?
Hey Babe, I am on topic. You presented some cut and past and acted like it was a fact that Kent has been found guilty of some crime by saying his cars were seized.
GUESS WHAT!
They gave the cars back. And whether you like it or not, it's part of the story.
So stop with the kiddie cut and paste and find out what really happened down there.
I often wonder what will they say when their children or friends (if they have any) bother to look up their posting history a few years from now. I could guess .. but I prefer not to soil myself with that.
"You have NO authority to decide this,"
And right you are. Your fate is in your hands. You're deciding your own fate. But remember that elitist fortress you're surrounding yourself with won't help. Eventually you will have to abandon it for safer lands. Back to Mother Europe, perhaps?
"and you should stick to worrying about your own future fate..."
I am.
Sidenote: I've been known for great visions. What I say above shall happen. You may not be a witness to it, but your children or children's childrens will.
I wish you luck on your journey.
Why are you so nasty to people who are Christians and believe the Bible?
Maybe you ought to law off the sauce for a while.
Exactly. It is so-called Christians who distort the Word of God (the Bible) for their own purposes who fragement the faith.
The Bible is clear for all those who are really interested in what God has to say.
Evolution is a lie just as the claim God sanctions homosexuality is a lie.
The is absolutely NOTHING in the Bible to support either of these.
Proverbs 20:1 ¶Wine is a mocker, strong drink is raging: and whosoever is deceived thereby is not wise.
And personally I have never cared too much for drunks. Especially people who drink in front of their kids.
You obviously didn't major in Biology...
I am an ex-evolutionists turned Christian.
Somehow I doubt that.
Evolution is an idiot theory designed to disprove the Bible.
Wrong, but thanks for playing. But just for fun, please explain in detail why it's an "idiot theory", and why the evidence for it is flawed (be specific, make sure you cover a lot of the lines of evidence).
It just astounds me that people could believe such a transparently idiotic theory as evolution. Monkeys become people.
Then perhaps you could explain what's "transparently idiotic" about the following (let's apply your high GPA to the task):
And:Background: Retroviruses reproduce by entering a cell of a host (like, say, a human), then embedding their own viral DNA into the cell's own DNA, which has the effect of adding a "recipe" for manufacturing more viruses to the cell's "instruction book". The cell then follows those instructions because it has no reason (or way) to "mistrust" the DNA instructions it contains. So the virus has converted the cell into a virus factory, and the new viruses leave the cell, and go find more cells to infect, etc.
However, every once in a while a virus's invasion plans don't function exactly as they should, and the virus's DNA (or portions of it) gets embedded into the cell's DNA in a "broken" manner. It's stuck into there, becoming part of the cell's DNA, but it's unable to produce new viruses. So there it remains, embedded in the DNA. If this happens in a regular body cell, it just remains there for life as a "fossil" of the past infection and goes to the grave with the individual it's stuck in. All of us almost certainly contain countless such relics of the past viral infections we've fought off.
However... By chance this sometimes happens to a special cell in the body, a gametocyte cell that's one of the ones responsible for making sperm in males and egg cells in females, and if so subsequent sperm/eggs produced by that cell will contain copies of the "fossil" virus, since now it's just a portion of the entire DNA package of the cell. And once in a blue moon such a sperm or egg is lucky enough to be one of the few which participate in fertilization and are used to produce a child -- who will now inherit copies of the "fossilized" viral DNA in every cell of his/her body, since all are copied from the DNA of the original modified sperm/egg.
So now the "fossilized" viral DNA sequence will be passed on to *their* children, and their children's children, and so on. Through a process called neutral genetic drift, given enough time (it happens faster in smaller populations than large) the "fossil" viral DNA will either be flushed out of the population eventually, *or* by luck of the draw end up in every member of the population X generations down the road. It all depends on a roll of the genetic dice.
Due to the hurdles, "fossil" retroviral DNA strings (known by the technical name of "endogenous retroviruses") don't end up ubiquitous in a species very often, but it provably *does* happen. In fact, the Human DNA project has identified literally *thousands* of such fossilized "relics" of long-ago ancestral infections in the human DNA.
And several features of these DNA relics can be used to demonstrate common descent, including their *location*. The reason is that retroviruses aren't very picky about where their DNA gets inserted into the host DNA. Even in an infection in a *single* individual, each infected cell has the retroviral DNA inserted into different locations than any other cell. Because the host DNA is so enormous (billions of basepairs in humans, for example), the odds of any retroviral insertion event matching the insertion location of any other insertion event are astronomically low. The only plausible mechanism by which two individuals could have retroviral DNA inserted into exactly the same location in their respective DNAs is if they inherited copies of that DNA from the same source -- a common ancestor.
Thus, shared endogenous retroviruses between, say, ape and man is almost irrefutable evidence that they descended from a common ancestor. *Unless* you want to suggest that they were created separately, and then a virus they were both susceptible to infected both a man and an ape in EXACTLY the same location in their DNAs (the odds of such a match by luck are literally on the order of 1,000,000,000,000 to 1...), *and* that the infections both happened in their gametocyte cells (combined odds on the order of 1,000,000 to 1) *and* that the one particular affected gametocyte is the one which produces the egg or sperm which is destined to produce an offspring (*HUGE* odds against), and *then* the resulting modified genome of the offspring becomes "fixed" in each respective population (1 out of population_size^squared)...
Then repeat that for *each* shared endogenous retrovirus (there are many) you'd like to claim was acquired independently and *not* from a shared ancestor...
Finally, you'd have to explain why, for say species A, B, and C, the pattern of shared same-location retroviruses is always *nested*, never *overlapped*. For example, all three will share some retroviruses, then A and B will both share several more, but if so then B *never* shares one with C that A doesn't also have (or at least remnants of).
In your "shared infection due to genetic similarities" suggestion, even leaving aside the near statistical impossibility of the infections leaving genetic "scars" in *exactly* the same locations in independent infections, one would expect to find cases of three species X, Y, and Z, where the degree of similarity was such that Y was "between" X and Z on some similarity scale, causing the same disease to befall X and Y but not Z, and another disease to affect Y and Z but not X. And yet, we don't find this in genetic markers. The markers are found in nested sequence, which is precisely what we would expect to see in cases of inheritance from common ancestry.
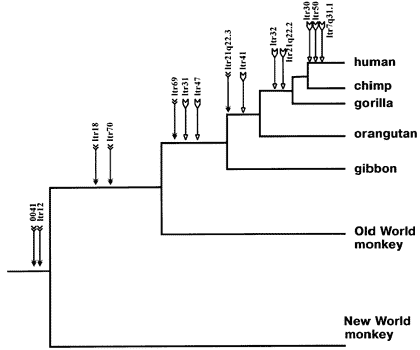
Here, for example, is an ancestry tree showing the pattern of shared same-location endogenous retroviruses of type HERV-K among primates:
This is just a partial list for illustration purposes -- there are many more.
Each labeled arrow on the chart shows an ERV shared in common by all the branches to the right, and *not* the branches that are "left-and-down". This is the pattern that common descent would make. And common descent is the *only* plausible explanation for it. Furthermore, similar findings tie together larger mammal groups into successively larger "superfamilies" of creatures all descended from a common ancestor.
Any presumption of independent acquisition is literally astronomically unlikely. And "God chose to put broken relics of viral infections that never actually happened into our DNA and line them up only in patterns that would provide incredibly strong evidence of common descent which hadn't actually happened" just strains credulity (not to mention would raise troubling questions about God's motives for such a misleading act).
Once again, the evidence for common descent -- as opposed to any other conceivable alternative explanation -- is clear and overwhelming.
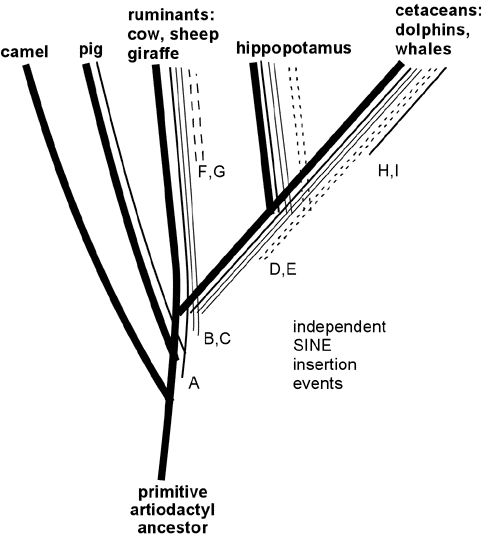
Wait, want more? Endogenous retroviruses are just *one* type of genetic "tag" that makes perfect sense evolutionary and *no* sense under any other scenario. In addition to ERV's, there are also similar arguments for the patterns across species of Protein functional redundancies, DNA coding redundancies, shared Processed pseudogenes, shared Transposons (including *several* independent varieties, such as SINEs and LINEs), shared redundant pseudogenes, etc. etc. Here, for example, is a small map of shared SINE events among various mammal groups:
Like ERV's, any scenario which suggests that these shared DNA features were acquired separately strains the laws of probability beyond the breaking point, but they make perfect sense from an evolutionary common-descent scenario. In the above data, it is clear that the only logical conclusion is that, for example, the cetaceans, hippos, and ruminants shared a common ancestor, in which SINE events B and C entered its DNA and then was passed on to its descendants, yet this occurred after the point in time where an earlier common ancestor had given rise both to that species, and to the lineage which later became pigs.
And this pattern (giving the *same* results) is repeated over and over and over again when various kinds of molecular evidence from DNA is examined in detail.
The molecular evidence for evolution and common descent is overwhelming. The only alternative is for creationists to deny the obvious and say, "well maybe God decided to set up all DNA in *only* ways that were consistent with an evolutionary result even though He'd have a lot more options open to him, even including parts which by every measure are useless and exactly mimic copy errors, ancient infections, stutters, and other garbage inherited from nonexistent shared ancestors"...
And since a couple times when I've posted this, clueless anti-evolutionists have mistaken their presumptions for reality and tried to declare that ERV insertion or analysis was just "speculative", here are a few relevant papers to chew on establishing their validity:
Human-specific integrations of the HERV-K endogenous retrovirus family
Endogenous retroviruses in the human genome sequence
Constructing primate phylogenies from ancient retrovirus sequences
Human L1 Retrotransposition: cis Preference versus trans Complementation
Identification, Phylogeny, and Evolution of Retroviral Elements Based on Their Envelope Genes
HERVd: database of human endogenous retroviruses
Long-term reinfection of the human genome by endogenous retroviruses
Insertional polymorphisms of full-length endogenous retroviruses in humans
Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans
The distribution of the endogenous retroviruses HERV-K113 and HERV-K115 in health and disease
A rare event of insertion polymorphism of a HERV-K LTR in the human genome
Demystified . . . Human endogenous retroviruses
Retroviral Diversity and Distribution in Vertebrates
Drosophila germline invasion by the endogenous retrovirus gypsy: involvement of the viral env gene
Genomic Organization of the Human Endogenous Retrovirus HERV-K(HML-2.HOM) (ERVK6) on Chromosome 7
Sequence variability, gene structure, and expression of full-length human endogenous retrovirus H
And, as usual, that's just the tiniest *tip* of the iceberg. My PubMed searches on endogenous retroviruses turned up over a *thousand* papers. These are just some of the more useful ones.
Humans have 23 pairs of chromosomes ---chimps and gorillas have 24 pairs. How many pairs of chromosomes did the "common ancestor" have? Was it 23 or 24 pairs? How do you "evolve" missing or added chromosomes ---that would happen all at one time.And:The common ancestor had 24 chromosomes.
If you look at the gene sequences, you'll find that Chromosome 2 in humans is pretty much just 2 shorter chimpanzee chromosomes pasted end-to-end, with perhaps a slight bit of lost overlap:
(H=Human, C=Chimpanzee, G=Gorilla, O=Orangutan)
Somewhere along the line, after humans split off from the other great apes, or during the split itself, there was an accidental fusion of two chromosomes, end-to-end. Where there used to be 24 chromosomes, now there were 23, but containing the same total genes, so other than a "repackaging", the DNA "instructions" remained the same.
If a chimpanzee gives birth to a creature with 23 chromosomes, that offspring isn't going to be a well-formed chimpanzee able to survive well.
It is if the same genes are present, which they would be in the case of a chromosome fusion.
Evolve would imply the genetic material changes little by little --not some big loss of two chromosomes at once but I don't see how they'd go away gene by gene.
Tacking two chromosomes together end-to-end is not a "big loss" of genes, and it really is a "little by little" change in the total genetic code. It's just been "regrouped" a bit. Instead of coming in 24 "packages", it's now contained in 23, but the contents are the same.
So how, you might ask, would the chromosomes from the first 23-chromosome "fused" individual match up with the 24 chromosomes from its mate when it tried to produce offspring? Very well, thanks for asking. The "top half" of the new extra-long Chromosome 2 would adhere to the original chromosome (call it "2p") from which it was formed, and likewise for the "bottom half" which would adhere to the other original shorter chromosome (call it "2q"). In the picture above, imagine the two chimp chromosomes sliding over to "match up" against the human chromosome. The chimp chromosomes would end up butting ends with each other, or slightly overlapping in a "kink", but chromosomes have overcome worse mismatches (just consider the XY pair in every human male -- the X and the Y chromosome are *very* different in shape, length, and structure, but they still pair up).
In fact, the "rubbing ends" of the matched-up chimp chromosomes, adhering to the double-long human-type chromosome, would be more likely to become fused together themselves.
For studies in which recent chromosome fusions have been discovered and found not to cause infertility, see:
Chromosomal heterozygosity and fertility in house mice (Mus musculus domesticus) from Northern Italy. Hauffe HC, Searle JB Department of Zoology, University of Oxford, Oxford OX1 3PS, United Kingdom. hauffe@novanet.itIn that last reference, the Przewalski horse, which has 33 chromosomes, and the domestic horse, with 32 chromosomes (due to a fusion), are able to mate and produce fertile offspring.An observed chromosome fusion: Hereditas 1998;129(2):177-80 A new centric fusion translocation in cattle: rob (13;19). Molteni L, De Giovanni-Macchi A, Succi G, Cremonesi F, Stacchezzini S, Di Meo GP, Iannuzzi L Institute of Animal Husbandry, Faculty of Agricultural Science, Milan, Italy.
J Reprod Fertil 1979 Nov;57(2):363-75 Cytogenetics and reproduction of sheep with multiple centric fusions (Robertsonian translocations). Bruere AN, Ellis PM
J Reprod Fertil Suppl 1975 Oct;(23):356-70 Cytogenetic studies of three equine hybrids. Chandley AC, Short RV, Allen WR.
Meanwhile, the question may be asked, how do we know that the human Chromosome 2 is actually the result of a chromsome fusion at/since a common ancestor, and not simply a matter of "different design"?
Well, if two chromsomes accidentally merged, there should be molecular remnants of the original chromosomal structures (while a chromosome designed from scratch would have no need for such leftover "train-wreck" pieces).
Ends of chromosomes have characteristic DNA base-pair sequences called "telomeres". And there are indeed remnants of telomeres at the point of presumed fusion on human Chromosome 2 (i.e., where the two ancestral ape chromosomes merged end-to-end). If I may crib from a web page:
Telomeres in humans have been shown to consist of head to tail repeats of the bases 5'TTAGGG running toward the end of the chromosome. Furthermore, there is a characteristic pattern of the base pairs in what is called the pre-telomeric region, the region just before the telomere. When the vicinity of chromosome 2 where the fusion is expected to occur (based on comparison to chimp chromosomes 2p and 2q) is examined, we see first sequences that are characteristic of the pre-telomeric region, then a section of telomeric sequences, and then another section of pre-telomeric sequences. Furthermore, in the telomeric section, it is observed that there is a point where instead of being arranged head to tail, the telomeric repeats suddenly reverse direction - becoming (CCCTAA)3' instead of 5'(TTAGGG), and the second pre-telomeric section is also the reverse of the first telomeric section. This pattern is precisely as predicted by a telomere to telomere fusion of the chimpanzee (ancestor) 2p and 2q chromosomes, and in precisely the expected location. Note that the CCCTAA sequence is the reversed complement of TTAGGG (C pairs with G, and T pairs with A).Another piece of evidence is that if human Chromosome 2 had formed by chromosome fusion in an ancestor instead of being designed "as is", it should have evidence of 2 centromeres (the "pinched waist" in the picture above -- chromosomes have centromeres to aid in cell division). A "designed" chromosome would need only 1 centromere. An accidentally "merged" chromosome would show evidence of the 2 centromeres from the two chromosomes it merged from (one from each). And indeed, as documented in (Avarello R, Pedicini A, Caiulo A, Zuffardi O, Fraccaro M, Evidence for an ancestral alphoid domain on the long arm of human chromosome 2. Hum Genet 1992 May;89(2):247-9), the functional centromere found on human Chromosome 2 lines up with the centromere of the chimp 2p chromosome, while there are non-functional remnants of the chimp 2q centromere at the expected location on the human chromosome.As an aside, the next time some creationist claims that there is "no evidence" for common ancestry or evolution, keep in mind that the sort of detailed "detective story" discussed above is repeated literally COUNTLESS times in the ordinary pursuit of scientific research and examination of biological and other types of evidence. Common ancestry and evolution is confirmed in bit and little ways over and over and over again. It's not just something that a couple of whacky anti-religionists dream up out of thin air and promulgate for no reason, as the creationists would have you believe.
[The poster known as Mr. LLLICHY wrote:] Here is that Vitamin C dataAnd:After discovering this same data on another thread along with more discussion than has appeared here (I've taken the liberty of pinging the participants of that discussion), I see what the "mystery" is supposed to be -- it's supposed be why did some sites have multiple mutations while (small) stretches of other sites had none? In other words, why do the mutations appear clustered?
(You know, it would really help if people explained their points and questions in more detail, instead of leaving people to guess what the poster was thinking...)
[LLLICHY wrote:] "U238" that decays thrice, pretty good trick when there is "U238" that does not decay at all in 50,000,000 years.
Actually, no site had mutations "thrice". Three different bases at a given site is only *two* mutations (one original base, plus two mutations from it to something else).
Here's the "mutation map" from the actual DNA data:
--1-12--1-1-1-1--------1112112--1---1-11-1--------1 ALL/nNo mutations ("-") in about half the sites, one mutation at several (17) sites, two mutations at three sites.The first thing to keep in mind that random processes tend to "cluster" more than people expect anyway. People expect "randomness" to "spread out" somewhat evenly, but instead it's usually more "clumped", for statistical reasons that would be a diversion to go into right now. So "that looks uneven" isn't always a good indication that something truly is non-random.
If you don't believe me on that, I wrote a program which made 23 mutations totally at random on a 51-site sequence, then repeated the process to see what different random outcomes would look like:
10 X$=STRING$(51,"-") 20 FOR I=1 TO 23 30 J%=INT(RND*51)+1 40 C$=MID$(X$,J%,1) 50 IF C$="-" THEN MID$(X$,J%,1)="1" ELSE MID$(X$,J%,1)=CHR$(ASC(C$)+1) 60 NEXT I 70 PRINT X$ 80 GOTO 10Yeah, it's BASIC, so sue me. Here's a typical screenful of the results:-21---1---2---111----2-----2-1121-------1---1--11-1 -1--1--21-11---1-1--1-1---1----1---21-11111---11--- 3-11---3-----1-----11-2-1---1--1----3--2---1--1---- ---1-1--22--1-1--2-2111--1-1111---1------1-------1- ---32----1-11-1-----1---2-231----1------1-----11--1 ----2---21--1---4----1-------------11-1--111-11-211 11--1-1---1-----1--1------1----3111--1----111-2-1-2 1112---1-3-1----1-1-----1-1------121--111-------1-1 -111121--1----1----1-1-1-1-11-2---1-1-------1-111-- -----------11-1---11-11--------21----12211--1---131 --1-211-1-1----21--11-1-2----1--1----11---11-----11 12---1-13------------2---21-21---11-1-1-1--2------- -----2-1---1-1----21--11-11-1---111-1--111-----2--1 -----1-----1-1-1-1---1-2----11-21-11--1-111---1-21- ---11--1-1-122-1-1-1--1-----2-1-1-1-------1-1---111 --2--11----2--1---12-2----1-1---1-1--1--12----1-1-1 -111-1-----1-1----------1-21111--1-2-11-11-1----11- 11-1--211-1221-----1--1-----11--1-2-1----------11-- -----1-12-11---2-1---11--1-2--1----11---111-1----11 11----1--12---12----1---31---1-11----2--1-11-1----- ---1--111-1--1-1-111----1-21----1-1-3---1------2--1 -2-11----1-1------1------2-1-1--111-111-1-1----1111 1--1--1-1---1-111111--2--1-1------112----2---11----Notice how oddly "clustered" most of them look, including one run which left a 13-site stretch "absolutely untouched", contrary to intuition (while having *4* mutations at a single site!)Frankly, I don't see anything in the real-life DNA mutation map which looks any different from these truly random runs. Random events tend to cluster more than people expect. That solves the "mystery" right there.
Also, there may be a selection factor -- the GLO gene is a *lot* bigger than this. One has to wonder if this small 51-bp section was presented just because it was the one that looked "least random". That would be a no-no, since one can always hand-select the most deviant subset out of larger sample in order to artificially skew the picture.
However, since there are some interesting evolutionary observations to be made, let's look at that DNA data again, slightly rearranged:
TAC CCC GTG GAG GTG CGC TTC ACT CGG GCG GAC GAC ATC CTG CTG AGC CCC PIG TAC CCC GTG GAG GTA CGC TTC ACT CGC GGG GAC GAC ATC CTG CTG AGC CCC BOS TAC CCC GTA GAG GTG CGC TTC ACC CGA GGC GAT GAC ATT CTG CTG AGC CCC RAT TAC CCC GTG GAG GTG CGC TTC ACC CGA GGT GAT GAC ATC CTG CTG AGC CCG MOUSE TAC CCT GTG GGG GTG CGC TTC ACC CGG GGG GAC GAC ATC CTG CTG AGC CCC GUIN PIG TAC CTG GTG GGG GTA CGC TTC ACC TGG AG* GAT GAC ATC CTA CTG AGC CCC HUMAN TAC CTG GTG GGG CTA CGC TTC ACC TGG AG* GAT GAC ATC CTA CTG AGC CCC CHIMPANZEE TAC CCG GTG GGG GTG CGC TTC ACC CAG AG* GAT GAC GTC CTA CTG AGC CCC ORANGUTAN TAA CCG GTG GGG GTG CGC TTC ACC CAA GG* GAT GAC ATC ATA CTG AGC CCC MACAQUEHere I've put spaces between codons, and clustered the closely-related species together: pig/cow as ungulates, rat/mouse for their obvious relationship, guinea pig right below them but separated because of the pseudogene nature of its GLO gene, then primates all in a group, with man's closest relative, the chimp, immediately below him, followed by the more distant orangutan, and the even more distant macaque. Also note that the top four have "working" GLO genes, and the bottom five have "broken" GLO pseudogenes.First, let's consider just the four species with working GLO genes. Evolution predicts that even over large periods of time, these genes will be "highly conserved", with natural selection weeding out mutations that could "break" the gene. Note that the mutations will still have occurred in individuals of the population, but natural selection will "discourage" that mutation from spreading into the general population.
And before we go any further, let's talk about the "universal genetic code". In all mammals (indeed, in almost all living organisms), each triplet of DNA sites cause a particular amino acid to be formed. The mapping of triplets (called "codons") to amino acids is as follows:
Second Position of Codon T C A G F
i
r
s
t
P
o
s
i
t
i
o
nT
TTT Phe [F] TTC Phe [F] TTA Leu [L] TTG Leu [L]
TCT Ser [S] TCC Ser [S] TCA Ser [S] TCG Ser [S]
TAT Tyr [Y] TAC Tyr [Y] TAA Ter [end] TAG Ter [end]
TGT Cys [C] TGC Cys [C] TGA Ter [end] TGG Trp [W]
T C A G T
h
i
r
d
P
o
s
i
t
i
o
nC
CTT Leu [L] CTC Leu [L] CTA Leu [L] CTG Leu [L]
CCT Pro [P] CCC Pro [P] CCA Pro [P] CCG Pro [P]
CAT His [H] CAC His [H] CAA Gln [Q] CAG Gln [Q]
CGT Arg [R] CGC Arg [R] CGA Arg [R] CGG Arg [R]
T C A G A
ATT Ile [I] ATC Ile [I] ATA Ile [I] ATG Met [M]
ACT Thr [T] ACC Thr [T] ACA Thr [T] ACG Thr [T]
AAT Asn [N] AAC Asn [N] AAA Lys [K] AAG Lys [K]
AGT Ser [S] AGC Ser [S] AGA Arg [R] AGG Arg [R]
T C A G G
GTT Val [V] GTC Val [V] GTA Val [V] GTG Val [V]
GCT Ala [A] GCC Ala [A] GCA Ala [A] GCG Ala [A]
GAT Asp [D] GAC Asp [D] GAA Glu [E] GAG Glu [E]
GGT Gly [G] GGC Gly [G] GGA Gly [G] GGG Gly [G]
T C A G (The above table imported from http://psyche.uthct.edu/shaun/SBlack/geneticd.html, which also has a nice introduction to the genetic code.)
Another version of the same table with nifty Java features and DNA database lookups can be found here.
The thing which is most relevant to the following discussion is the fact that most of the genetic codes are "redundant" -- more than one codon (triplet) encodes to exactly the same amino acid. This means that even in genes which are required for the organism, certain basepair mutations make absolutely no difference if the change is from one codon which maps into amino acid X to another codon which still maps into amino acid X. (This fact allows certain kinds of evolutionary "tracers" to be "read" from the DNA, as described here).
Now back to our DNA data. The redundancy in the genetic code means that some basepair sites will have more "degrees of freedom" than others (i.e., ways in which they can mutate without disrupting the gene's biological function in any way). Let's look at the four species with working GLO genes again:
TAC CCC GTG GAG GTG CGC TTC ACT CGG GCG GAC GAC ATC CTG CTG AGC CCC PIG TAC CCC GTG GAG GTA CGC TTC ACT CGC GGG GAC GAC ATC CTG CTG AGC CCC BOS TAC CCC GTA GAG GTG CGC TTC ACC CGA GGC GAT GAC ATT CTG CTG AGC CCC RAT TAC CCC GTG GAG GTG CGC TTC ACC CGA GGT GAT GAC ATC CTG CTG AGC CCG MOUSE T T T A T A T T T A T C C T T T T T T T T A A A A A C A A A A A G C G G G G G C C C --- --- --1 --- --1 --- --- --1 --2 -12 --1 --- --1 --- --- --- --1Under each site of the mouse DNA, I've listed the "alternative" bases which could be be substituted for the mouse base at that site WITHOUT ALTERING THE GENE'S FUNCTION (because of genetic code redundancy). And under that I show the "mutation map" of just those four species.Note that most of the "alternative" bases are in the third base of each codon, *and* that this is where all but one of the mutations have appeared. This is because these were the sites which were "free" to mutate in the way they did, because the mutation was genetically neutral. That doesn't mean that the first and second sites of each codon were immune from mutation, it's just that when mutations did occur at those sites, natural selection weeded them out quickly because they most likely "broke" the GLO gene for the individuals which received that mutuation. What we see above is the results after natural selection has already "filtered" the undesirable mutations and left the ones which "do no harm".
Additionally, the two sites which have mutated twice (i.e. have a "2" in the mutation map) are ones which had more "allowable" mutations. Also note that the sites which had the fewest allowable alternatives (only one alternate letter allowed) didn't have any mutations fix at those sites, which is unsurprising since a "safe" mutation would be less likely to occur there versus a site that "allowed" two or three alternatives.
All this is as predicted by evolutionary theory, you'll note.
It also explains the one anomoly of the original mutation map, which is that the mutation counts do tend to be higher at the third base of a codon.
However... What about the one exception? The pig DNA has had one mutation at a site which does not encode to exactly the same amino acid (which is the case for *all* the other ones). In the pig DNA, the GGG codon (mapping to Glycine) has changed to a GCG codon (mapping to Alanine). What's up with that? Well, one of two things. First and most likely, just as base values in codons have a built-in redundancy, so do the amino acids which make up the proteins which result from the DNA templates. In other words, certain amino acids can be substituted for other ones at some sites in given proteins without making any functional difference. (This "protein functional redundancy" also has implications for "evolutionary tracer" analysis, see here.) That may well be the case for Alanine versus Glycine in the GLO protein, but I'm not enough of a biochemist to be able to say. The other option is that it *does* make some difference in the function of the pig GLO protein, but not enough to "break" the vitamin-C synthesis (as proven by the fact that pigs *can* synthesize vitamin C). So one way or another, it's not a deal-breaker even though pig GLO will not be 100% identical to cow/mouse/rat GLO. It's yet another "allowable" mutation.
More interesting evolutionary observations: The number of mutational differences between pig/cow is 3, the number between mouse/rat is 4, and the difference between rat/cow is 7 -- all roughly as one would expect from the evolutionary relatedness of these animals (cows/pigs and rats/mice are each closer to each other than the rodents are to the ungulates).
Now let's take a close look at the guinea pig:
TAC CCT GTG GGG GTG CGC TTC ACC CGG GGG GAC GAC ATC CTG CTG AGC CCC GUIN PIG --- --1 --- -1- --- --- --- --- --1 --1 --1 --- --- --- --- --- ---The "mutation map" under the guinea pig DNA is compared to the mouse DNA. Fascinating: Note that four of the five mutations are in the third base of a codon, *and* are of the type "allowed" by the genetic code redundancy. This indicates strongly that most of the evolutionary divergence between guinea pigs and mice likely occurred while the guinea pig's ancestors still had a working GLO gene. This is the sort of prediction implied by the evolutionary theory which could be cross-checked by further research of various types, and if verified, would be yet further confirmation that evolutionary theory is likely correct. So far, evolutionary theory has been subjected to literally countless tests like this, large and small, and the vast majority of results have confirmed the evolutionary prediction. This track record is hard to explain if evolution is an invalid theory, as some assert...Finally, let's look over the primate DNA and mutation map (relative to each other):
TAC CTG GTG GGG GTA CGC TTC ACC TGG AG* GAT GAC ATC CTA CTG AGC CCC HUMAN TAC CTG GTG GGG CTA CGC TTC ACC TGG AG* GAT GAC ATC CTA CTG AGC CCC CHIMPANZEE TAC CCG GTG GGG GTG CGC TTC ACC CAG AG* GAT GAC GTC CTA CTG AGC CCC ORANGUTAN TAA CCG GTG GGG GTG CGC TTC ACC CAA GG* GAT GAC ATC ATA CTG AGC CCC MACAQUE --1 -1- --- --- 1-1 --- --- --- 111 1-- --- --- 1-- 1-- --- --- ---Evolutionary theory predicts that because the GLO gene is "broken" in primates (i.e. is a pseudogene), mutations in it are highly likely to be neutral (i.e., make no difference, since it can't get much more broken), and thus mutations are just as likely to accumulate at any site as any other. Is that what we see? Yup. There's no obvious pattern to the mutations between primates in the above mutation map, and unlike the pig/cow/mouse/rat mutation map, the mutations aren't predominantly at the "safer" third base of a codon, nor of a type that would be "safe". In fact, one base has vanished entirely, but no biggie, the gene's already broken.Also, although primates share a more recent common ancestor than cows/pigs/mice/rats, note that they've already racked up almost as many relative mutations as the cow/pig/mouse/rat DNA. This too is just as evolutionary theory predicts, because many mutations in a functional gene (GLO in this case) will be "non-safe" and weeded out by natural selection, making for a slower mutation fixation rate overall than in a pseudogene (as GLO is in primates) where natural selection doesn't "care" about the vast majority of mutations since *most* are neutral. So pseudogenes accumulate mutations faster than functional genes (even though rate of mutation *occurence* in both are likely the same).
Finally, note that there are ZERO mutational differences between the human DNA and the chimpanzee DNA, our nearest living relative.
I also see some interesting implications in the DNA sequences concerning which specific mutation fixed during what branch of the common-descent evolutionary tree for all the species represented, but reconstructing that would not only take another couple hours, at least, but would be a major bear to code in HTML, since I'd have to draw trees with annotations on the nodes... Bleugh.
In any case, I hope I've clarified some of the methods by which biologists find countless confirmations of evolution in DNA data. This is just a "baby" example, and to be more statistically valid would have to be done over much vaster sections of DNA sequences, but my intent was to demonstrate some of the concepts.
And if such a small amount of DNA as this can make small confirmations of evolutionary predictions, imagine the amount of confirmation from billion-basepair DNA data from each species compared across thousands of species... The amount of confirmatory discoveries for evolution from DNA analysis has already been vast, and promises to only grow in the future. For an overview of some of the different lines of evidence being studied, see The Journal of Molecular Evolution -- abstracts of all articles, current and back issues, can be browsed free online.
And:Where exactly is the "missing" transition in the following sequence? It looks pretty complete and gradual to me -- certainly there's no sudden "jump", no discontinuity, no pair between which a creationist would have any trouble dismissing such a small amount of change as "just microevolution", "just variation within a kind":
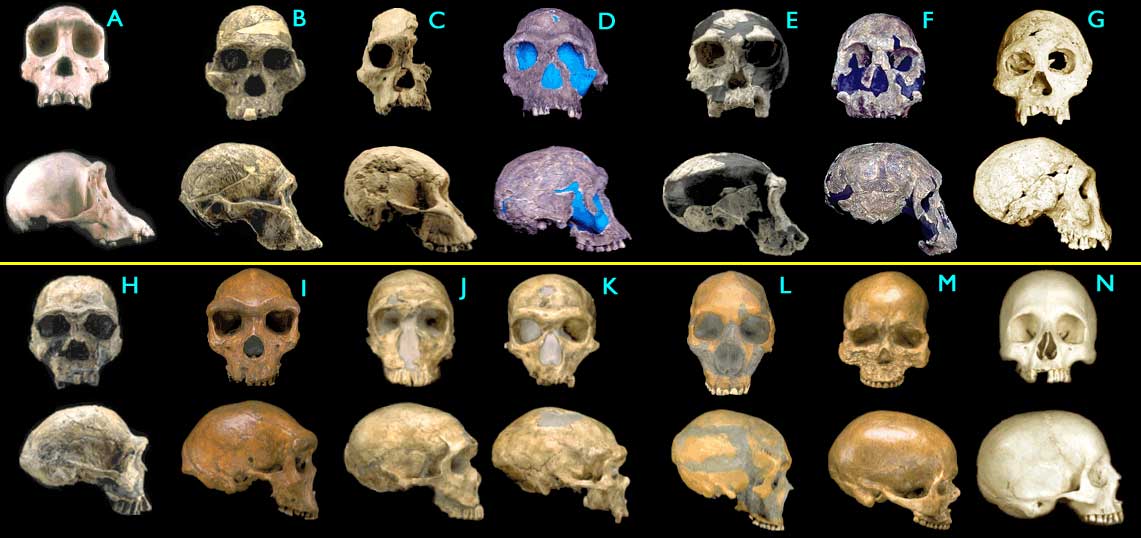
(The above is from 29 Evidences for Macroevolution -- Part 1: The Unique Universal Phylogenetic Tree)Figure 1.4.4. Fossil hominid skulls. Some of the figures have been modified for ease of comparison (only left-right mirroring or removal of a jawbone). (Images © 2000 Smithsonian Institution.)
- (A) Pan troglodytes, chimpanzee, modern
- (B) Australopithecus africanus, STS 5, 2.6 My
- (C) Australopithecus africanus, STS 71, 2.5 My
- (D) Homo habilis, KNM-ER 1813, 1.9 My
- (E) Homo habilis, OH24, 1.8 My
- (F) Homo rudolfensis, KNM-ER 1470, 1.8 My
- (G) Homo erectus, Dmanisi cranium D2700, 1.75 My
- (H) Homo ergaster (early H. erectus), KNM-ER 3733, 1.75 My
- (I) Homo heidelbergensis, "Rhodesia man," 300,000 - 125,000 y
- (J) Homo sapiens neanderthalensis, La Ferrassie 1, 70,000 y
- (K) Homo sapiens neanderthalensis, La Chappelle-aux-Saints, 60,000 y
- (L) Homo sapiens neanderthalensis, Le Moustier, 45,000 y
- (M) Homo sapiens sapiens, Cro-Magnon I, 30,000 y
- (N) Homo sapiens sapiens, modern
And:Fossil Hominids: The Evidence for Human Evolution.
29 Evidences for Macroevolution -- Part 1: The Unique Universal Phylogenetic Tree
Analysis of the human Alu Ye lineage
Molecular and temporal characteristics of human retropseudogenes.
Serine hydroxymethyltransferase pseudogene, SHMT-ps1: a unique genetic marker of the order primates
Insertions and duplications of mtDNA in the nuclear genomes of Old World monkeys and hominoids
Fixation times of retroposons in the ribosomal DNA spacer of human and other primates
The emergence of new DNA repeats and the divergence of primates
Genetic diversity at class II DRB loci of the primate MHC
Structure and evolution of human and African ape rDNA pseudogenes
Accelerated Evolution of the ASPM Gene Controlling Brain Size Begins Prior to Human Brain ExpansionEtc., Etc., Etc. How many more thousands of papers would you like to see covering the common ancestry that you think is "transparently idiotic"?Abstract: Primary microcephaly (MCPH) is a neurodevelopmental disorder characterized by global reduction in cerebral cortical volume. The microcephalic brain has a volume comparable to that of early hominids, raising the possibility that some MCPH genes may have been evolutionary targets in the expansion of the cerebral cortex in mammals and especially primates. Mutations in ASPM, which encodes the human homologue of a fly protein essential for spindle function, are the most common known cause of MCPH. Here we have isolated large genomic clones containing the complete ASPM gene, including promoter regions and introns, from chimpanzee, gorilla, orangutan, and rhesus macaque by transformation-associated recombination cloning in yeast. We have sequenced these clones and show that whereas much of the sequence of ASPM is substantially conserved among primates, specific segments are subject to high Ka/Ks ratios (nonsynonymous/synonymous DNA changes) consistent with strong positive selection for evolutionary change. The ASPM gene sequence shows accelerated evolution in the African hominoid clade, and this precedes hominid brain expansion by several million years. Gorilla and human lineages show particularly accelerated evolution in the IQ domain of ASPM. Moreover, ASPM regions under positive selection in primates are also the most highly diverged regions between primates and nonprimate mammals. We report the first direct application of TAR cloning technology to the study of human evolution. Our data suggest that evolutionary selection of specific segments of the ASPM sequence strongly relates to differences in cerebral cortical size.Identification of paralogous HERV-K LTRs on human chromosomes 3, 4, 7 and 11 in regions containing clusters of olfactory receptor genesAbstract: A locus harboring a human endogenous retroviral LTR (long terminal repeat) was mapped on the short arm of human chromosome 7 (7p22), and its evolutionary history was investigated. Sequences of two human genome fragments that were homologous to the LTR-flanking sequences were found in human genome databases: (1) an LTR-containing DNA fragment from region 3p13 of the human genome, which includes clusters of olfactory receptor genes and pseudogenes; and (2) a fragment of region 21q22.1 lacking LTR sequences. PCR analysis demonstrated that LTRs with highly homologous flanking sequences could be found in the genomes of human, chimp, gorilla, and orangutan, but were absent from the genomes of gibbon and New World monkeys. A PCR assay with a primer set corresponding to the sequence from human Chr 3 allowed us to detect LTR-containing paralogous sequences on human chromosomes 3, 4, 7, and 11. The divergence times for the LTR-flanking sequences on chromosomes 3 and 7, and the paralogous sequence on chromosome 21, were evaluated and used to reconstruct the order of duplication events and retroviral insertions. (1) An initial duplication event that occurred 14-17 Mya and before LTR insertion - produced two loci, one corresponding to that located on Chr 21, while the second was the ancestor of the loci on chromosomes 3 and 7. (2) Insertion of the LTR (most probably as a provirus) into this ancestral locus took place 13 Mya. (3) Duplication of the LTR-containing ancestral locus occurred 11 Mya, forming the paralogous modern loci on Chr 3 and 7.Birth and adaptive evolution of a hominoid gene that supports high neurotransmitter fluxAbstract: The enzyme glutamate dehydrogenase (GDH) is important for recycling the chief excitatory neurotransmitter, glutamate, during neurotransmission. Human GDH exists in housekeeping and brain-specific isotypes encoded by the genes GLUD1 and GLUD2, respectively. Here we show that GLUD2 originated by retroposition from GLUD1 in the hominoid ancestor less than 23 million years ago. The amino acid changes responsible for the unique brain-specific properties of the enzyme derived from GLUD2 occurred during a period of positive selection after the duplication event.A uniquely human consequence of domain-specific functional adaptation in a sialic acid–binding receptorAbstract: Most mammalian cell surfaces display two major sialic acids (Sias), N-acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). Humans lack Neu5Gc due to a mutation in CMP-Neu5Ac hydroxylase, which occurred after evolutionary divergence from great apes. We describe an apparent consequence of human Neu5Gc loss: domain-specific functional adaptation of Siglec-9, a member of the family of sialic acid–binding receptors of innate immune cells designated the CD33-related Siglecs (CD33rSiglecs). Binding studies on recombinant human Siglec-9 show recognition of both Neu5Ac and Neu5Gc. In striking contrast, chimpanzee and gorilla Siglec-9 strongly prefer binding Neu5Gc. Simultaneous probing of multiple endogenous CD33rSiglecs on circulating blood cells of human, chimp, or gorilla suggests that the binding differences observed for Siglec-9 are representative of multiple CD33rSiglecs. We conclude that Neu5Ac-binding ability of at least some human CD33rSiglecs is a derived state selected for following loss of Neu5Gc in the hominid lineage. These data also indicate that endogenous Sias (rather than surface Sias of bacterial pathogens) are the functional ligands of CD33rSiglecs and suggest that the endogenous Sia landscape is the major factor directing evolution of CD33rSiglec binding specificity. Exon-1-encoded Sia-recognizing domains of human and ape Siglec-9 share only 93–95% amino acid identity. In contrast, the immediately adjacent intron and exon 2 have the 98–100% identity typically observed among these species. Together, our findings suggest ongoing adaptive evolution specific to the Sia-binding domain, possibly of an episodic nature. Such domain-specific divergences should also be considered in upcoming comparisons of human and chimpanzee genomes.Lineage-Specific Gene Duplication and Loss in Human and Great Ape EvolutionAbstract: Given that gene duplication is a major driving force of evolutionary change and the key mechanism underlying the emergence of new genes and biological processes, this study sought to use a novel genome-wide approach to identify genes that have undergone lineage-specific duplications or contractions among several hominoid lineages. Interspecies cDNA array-based comparative genomic hybridization was used to individually compare copy number variation for 39,711 cDNAs, representing 29,619 human genes, across five hominoid species, including human. We identified 1,005 genes, either as isolated genes or in clusters positionally biased toward rearrangement-prone genomic regions, that produced relative hybridization signals unique to one or more of the hominoid lineages. Measured as a function of the evolutionary age of each lineage, genes showing copy number expansions were most pronounced in human (134) and include a number of genes thought to be involved in the structure and function of the brain. This work represents, to our knowledge, the first genome-wide gene-based survey of gene duplication across hominoid species. The genes identified here likely represent a significant majority of the major gene copy number changes that have occurred over the past 15 million years of human and great ape evolution and are likely to underlie some of the key phenotypic characteristics that distinguish these species.Sequence Variation Within the Fragile X LocusAbstract: The human genome provides a reference sequence, which is a template for resequencing studies that aim to discover and interpret the record of common ancestry that exists in extant genomes. To understand the nature and pattern of variation and linkage disequilibrium comprising this history, we present a study of ~31 kb spanning an ~70 kb region of FMR1, sequenced in a sample of 20 humans (worldwide sample) and four great apes (chimp, bonobo, and gorilla). Twenty-five polymorphic sites and two insertion/deletions, distributed in 11 unique haplotypes, were identified among humans. Africans are the only geographic group that do not share any haplotypes with other groups. Parsimony analysis reveals two main clades and suggests that the four major human geographic groups are distributed throughout the phylogenetic tree and within each major clade. An African sample appears to be most closely related to the common ancestor shared with the three other geographic groups. Nucleotide diversity, [pi], for this sample is 2.63 ± 6.28 × 10-4. The mutation rate, [mu], is 6.48 × 10-10 per base pair per year, giving an ancestral population size of ~6200 and a time to the most recent common ancestor of ~320,000 ± 72,000 per base pair per year. Linkage disequilibrium (LD) at the FMR1 locus, evaluated by conventional LD analysis and by the length of segment shared between any two chromosomes, is extensive across the region.Structural and evolutionary analysis of the two chimpanzee alpha-globin mRNAsAbstract: Two distinct alpha-globin mRNAs were detected in chimpanzee reticulocyte mRNA using a primer extension assay. DNA copies of these two mRNAs were cloned in the bacterial plasmid pBR322, and their sequence was determined. The two alpha-globin mRNAs have obvious structural homology to the two human alpha-globin mRNAs, alpha 1 and alpha 2. Comparison of the two chimpanzee alpha-globin mRNAs to each other and to their corresponding human counterparts revealed evidence of a recent gene conversion in the human alpha-globin complex and a marked heterogeneity in the rate of structural divergence within the alpha-globin gene.Differential Alu Mobilization and Polymorphism Among the Human and Chimpanzee LineagesAbstract: Alu elements are primate-specific members of the SINE (short interspersed element) retroposon family, which comprise10% of the human genome. Here we report the first chromosomal-level comparison examining the Alu retroposition dynamics following the divergence of humans and chimpanzees. We find a twofold increase in Alu insertions in humans in comparison to the common chimpanzee (Pan troglodytes). The genomic diversity (polymorphism for presence or absence of the Alu insertion) associated with these inserts indicates that, analogous to recent nucleotide diversity studies, the level of chimpanzee Alu diversity is
You might be claiming that you are not drunk and that might be true, but you are posting things to me that make no sense at all and have nothing at all to do with anything I have said to you on this thread.
I took only a brief glance at the link in your post #884, and it was bizarre...well, you know, it was like this also, during the whole Terri Shiavo incident...
During that time, I got called a Nazi, because I simply asked the question, if it was considered a sin by the Catholic Church to be cremated...people said a lot of things during that time also, which were truly bizarre...
I read it then and re-read again now. The footnote does NOT come with the offer. It was added by whoever created the page - ie, Kent Hovind's foes.
Wrong. Here is Hovind's offer. The footnote is his, and it makes the offer a complete hoax.
In case you were wondering, you have been hoaxed.
It says to me that you're wrong.
Proverbs 20:1 ¶Wine is a mocker, strong drink is raging: and whosoever is deceived thereby is not wise.
I guess you think Paul was an evil person for suggesting wine for stomach problems and of course Jesus was truly wicked for turning water into wine.
This confirms something I had already suspected.
I don't know a soul posting on this thread. But my question is why do you think it's ok for you to disagree with people and it's ok, but let a Christian disagree with someone and somehow that makes them "bad" in your book.
I guess earnestly contending isn't in your bible.
I personally abhor those that would actually drink booze in front of innocent children.
Disclaimer: Opinions posted on Free Republic are those of the individual posters and do not necessarily represent the opinion of Free Republic or its management. All materials posted herein are protected by copyright law and the exemption for fair use of copyrighted works.